Leaving Community
Are you sure you want to leave this community? Leaving the community will revoke any permissions you have been granted in this community.
TIE2 - Overview
Challenge timeframe: Sep 01, 2017 to Dec 01, 2017
Angiopoietin-1 receptor (TIE2) is composed of an extracellular domain for ligand binding, a transmembrane domain, and a cytoplasmic tyrosine kinase domain. For this challenge, we focus on the cytoplasmic kinase domain which is a tyrosine kinase involved in tumor-induced angiogenesis [1, 2].
| 2017-11-19 | TIE2 Deadline - Dec 1, 2017 23:59PST |
- TIE2_target_D3R_GC3.fasta: Protein sequence file of the TIE2 kinase domain construct used in the Kd experiments.
- TIE2_score_compounds_D3R_GC3.csv: CSV file of 18 compound and their corresponding SMILES string, target, and subchallenge name.
- TIE2_FESet1_compounds_D3R_GC3.csv: CSV file of 4 compounds and SMILES strings selected from TIE2_score_compounds_D3R_GC3.csv for explicit-solvent relative or absolute free energy calculations.
- TIE2_FESet2_compounds_D3R_GC3.csv: CSV file of 6 compounds and SMILES strings selected from TIE2_score_compounds_D3R_GC3.csv for explicit-solvent relative or absolute free energy calculations.
Note: No attempt was made to set appropriate starting conformations or optimal protonation or tautomer states for the ligands, or to generate alternative tautomer states. It is up to you to choose and set these states for your calculations.
Template and Example packet: TIE2_Submission_examples_and_templates.zip
General instructions on what to include in each subchallenge component can be found at https://drugdesigndata.org/about/grand-challenge-3-submission-instructions.
This packet includes "templates" and "examples" folders.
The "template" folder includes files where values have been removed.
The "examples" folder includes complete submissions that passed validation:
- scorestructure.tgz
- scoreligand.tgz
- freeenergy2.tgz
- freeenergy1.tgz
Note: the data in the example TGZ files are merely for demonstration purposes and hold no scientific value.
General Information on Kinases
Protein kinases
The protein kinases are a family of enzymes that are involved in signal transduction pathways in cell biology. Due to their roles in various diseases, protein kinases have become attractive drug targets [3].
Architecture of active vs inactive protein kinases
Protein kinases have flexible ligand-binding domains that can result in induced-fit effects. Changes in binding site conformation involve multiple structural features: the glycine-rich loop, the activation loop, the catalytic loop, the P+1 loop, the αC-helix, and the DFG motif. Different combinations of these features' structural arrangements, along with the phosphorylation state of the activation loop, can result in an active or inactive form of the kinase, making it difficult to distinguish between the two forms. However, it is possible to determine whether a kinase is active or inactive by identifying the assembly pattern of a spatially conserved hydrophobic motif, termed the regulatory spine. In the active form, the regulatory spine has a near-linear arrangement, and in the inactive form, it adopts distorted arrangements with distinct inactive conformations. Another structural determinant of protein activation is the conformation of the DFG motif, which is capable of sterically blocking the binding of ATP. When the DFG motif is in an out conformation ("DFG-out"), the kinase is in its inactive form. On the other hand, the "DFG-in" conformation can still exhibit an active or inactive form of the kinase. In most cases, phosphorylation of the activation loop can also be an indication of kinase activation [4].
Kinase inhibitors
Small molecule inhibitors can bind to a wide range of kinase conformations that span several active and inactive forms. Kinase inhibitors are classified into several types, depending on binding pocket location or structural variation within the orthosteric binding site [3]:
- Type I inhibitors bind to the DFG-in active conformation,
- Type II inhibitors bind to the DFG-out inactive conformation,
- Type I1/2 inhibitors bind in the DFG-in inactive conformation but extend into the back pockets,
- Type III inhibitors bind next to the ATP-binding pocket,
- Type IV inhibitors are allosteric inhibitors that target protein kinases distal to the ATP binding pocket,
- Type V bisubstrate and bivalent inhibitors combine different inhibitor classes.
Inhibitor classifications are sometimes ambiguous because the kinase conformation is flexible and inhibitors are not necessarily conformationally selective.
Additional Information
- Most assays are performed using kinases just as they are expressed, and in the absence of ATP. Kinases expressed using bacterial phage lysates are thought to be in the inactive state, whereas kinases expressed in mammalian cells are thought to have a higher chance of being in an active state.
- The kinases for which the phosphorylated state is NOT specified (all datasets other than ABL1) can be in either an activated or inactivated state.
| Target | Expression system |
|---|---|
| VEGFR2 | Mammalian |
| JAK2 | Mammalian |
| p38-α | Bacterial |
| TIE2 | Bacterial |
| ABL1 | Mammalian |
Kinase Binding assay conditions
Kinase assays
The assays used here were carried out as previously described [5].
For most assays, kinase-tagged T7 phage strains were prepared in an E. coli host derived from the BL21 strain. E. coli were grown to log-phase and infected with T7 phage and incubated with shaking at 32°C until lysis. The lysates were centrifuged and filtered to remove cell debris. The remaining kinases were produced in HEK-293 cells and subsequently tagged with DNA for qPCR detection. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for kinase assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce non-specific binding. Binding reactions were assembled by combining kinases, liganded affinity beads, and test compounds in 1x binding buffer (20% SeaBlock, 0.17x PBS, 0.05% Tween 20, 6 mM DTT). Test compounds were prepared as 111X stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements were distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions were performed in polypropylene 384-well plates. Each was a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (1x PBS, 0.05% Tween 20). The beads were then re-suspended in elution buffer (1x PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR (see above)..
Compound Handling
An 11-point 3-fold serial dilution of each test compound was prepared in 100% DMSO at 100x final test concentration and subsequently diluted to 1x in the assay (final DMSO concentration = 1%). Most Kds were determined using a compound top concentration = 30,000 nM. If the initial Kd determined was < 0.5 nM (the lowest concentration tested), the measurement was repeated with a serial dilution starting at a lower top concentration. A Kd value reported as 40,000 nM indicates that the Kd was determined to be >30,000 nM.
Binding Constants (Kds)
Binding constants (Kds) were calculated with a standard dose-response curve using the Hill equation.
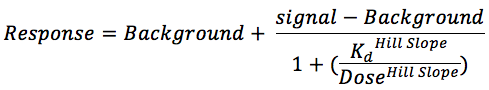
The Hill Slope was set to -1.
Curves were fitted using a non-linear least square fit with the Levenberg-Marquardt algorithm.
References:
1. Barton WA, Tzvetkova-Robev D, Miranda EP, et al (2006) Crystal structures of the Tie2 receptor ectodomain and the angiopoietin-2-Tie2 complex. Nat Struct Mol Biol 13:524-532. doi: 10.1038/nsmb1101
2. Pan P, Sun H, Liu H, et al (2016) In Silico Exploration for Novel Type-I Inhibitors of Tie-2/TEK: The Performance of Different Selection Strategy in Selecting Virtual Screening Candidates. Sci Rep. doi: 10.1038/srep37628
3. Roskoski R (2016) Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res 103:26-48. doi: 10.1016/j.phrs.2015.10.021
4. Taylor SS, Kornev AP (2011) Protein Kinases: Evolution of Dynamic Regulatory Proteins. Trends Biochem Sci 36:65-77. doi: 10.1016/j.tibs.2010.09.006
5. Fabian MA, Biggs WH, Treiber DK, et al (2005) A small molecule?kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23:329-336. doi: 10.1038/nbt1068
TIE2 - Data Download
Challenge timeframe: Sep 01, 2017 to Dec 01, 2017
TIE2 - Submissions
Challenge timeframe: Sep 01, 2017 to Dec 01, 2017
Please join the challenge and Login.
TIE2 - Evaluation Results
Challenge timeframe: Sep 01, 2017 to Dec 01, 2017
Evaluation Results
Overviews
Last updated April 9, 2018Pose Prediction
Affinity Rankings
This section presents metrics of the ability of the predictions to correctly rank ligands by affinity. The rankings were evaluated in terms of the Kendall's τ, Spearman's ρ, and estimated binding energies for the free energy sets were additionally evaluated in terms of centered root-mean-square error (RMSEc, kcal/mol) and Pearson's r. Uncertainties in these statistics (e.g., Kendall's τ Errors in the table) were obtained by recomputing them in 10,000 rounds of resampling with replacement, where, in each sample, the experimental IC50 or Kd data were randomly modified based on the experimental uncertainties. Experimental uncertainties are added to the free energy, ΔG, as a random offset δG drawn from a Gaussian distribution of mean zero and standard deviation RTln(Ierr). In this evaluation, the value of Ierr was set to 2.5.
For the kinases, a number of experimental Kd values were reported as ≥10 µM, making them difficult to include in standard metrics of ranking accuracy, so these cases were excluded from these affinity ranking assessments. However, they are included in the Active/Inactive Classification assessments, below.
Active / Inactive Classification
Affinity Rankings for Cocrystalized Ligands
User Submissions
GC3_CatS1A.tgz (508M)
GC3_CatS1B.tgz (184M)
GC3_CatS2.tgz (554M)
GC3_vegfr2_submissions.tgz (179M)
GC3_tie2_submissions.tgz (7.7M)
GC3_p38a_submissions.tgz (141M)
GC3_jak2sc2_submissions.tgz (210M)
GC3_jak2sc3_submissions.tgz (63M)
GC3_abl1_submissions.tgz (108M)
All scripts used to evaluate submissions are publicly available on Github.
(partials) indicates submissions that do not include the full set of predictions
About
Welcome to the Drug Design Data Resource Community. D3R is funded in part by NIH grant 1U01GM111528 from the National Institute of General Medical Sciences